![]()
![]()
![]()
![]()
| |
|
|
 Quality Management, Regulatory Affairs & Communication, Stress Management and Audit Psychology
Quality Management, Regulatory Affairs & Communication, Stress Management and Audit Psychology
Keywords: Medical Devices / Pharma Project Management (Europe, USA, Japan), Quality Management, GMP Guidelines and Legislation, e.g., AMWHV, EU-GMP Guidelines I, II, IV (ATMP) & Annexes, ICH Guidelines and FDA/USA 21 CFR 4, 11, 210/211, 820; MEDDEV / MDCG, MPG / MPDG, MDD, 93/42/EWG, MDR/IVDR 2017/745-746; MDSAP); Usability (Usability / Human Factors and Usability Engineering acc. to IEC 62366 / FDA CDRH); DIN EN ISO 13485:2012/2016; Audits / Inspections e.g. FDA; Regulatory Affairs, e.g., Medical Devices (510(k), former STED Format), Pharma CMC Part – Module 3, Combination Products / Companion Products, Phyto-Pharmaceuticals, e.g., Registration in Germany Europe, USA, Japan, Korea, Canada, Australia, Brazil); Work- and Organizational Psychology, Communication, Stress-Management, Audit Psychology - audit preparation of personnel, In-House Training & Lecturer for Quality Management.
For more information, please click on the following topics:
Consulting and hands-on support, e.g., issue or review of documents like quality manuals, PQRs / APRs according to EU-GMP Guideline part I - 1.10 (drugs) or part II - 2.6 (APIs) and ICH Q7 / 21 CFR 211, SOPs or forms, performance of GxP audits and gap-analyses, with the objective to establish a status of "optimum compliance" with legal requirements (e.g., EU / US GMP; GDP Guideline 2013/C 343/01), EU-directives and ISO standards (e.g., 9000 et seqq., 17025, 13485), which brings an added value to the quality and production processes --- quality management based on scientific understanding, and risk-based assessments that fits pragmatically and applicably into the existing system (e.g., people, stakeholder, corporate culture), considering the cost-benefit ratio.
In addition to the German national legislation AMG / AMWHV | MPG, requirements of, e.g., the US cGMP (e.g., 21 CFR Parts 210, 211, 820 QSR, 11, 4 [Final Guidance]), the ICH Quality Guidelines (e.g., Q7, Q9(R1) [2023], Q10), the harmonized ISO standards, the MDD / MDR or IVDR and the quality factors according to the 9 chapters of the EU GMP Guideline Part I, as well as Annexes 1 - 21 and if applicable Part IV (ATMP) are considered:
Chapter 1: Pharmaceutical Quality System
Chapter 2: Personnel
Chapter 3: Premise and Equipment
Chapter 4: Documentation
Chapter 5: Production
Chapter 6: Quality Control
Chapter 7: On Outsourced activities
Chapter 8: Complaints and Product Recall
Chapter 9: Self-Inspection
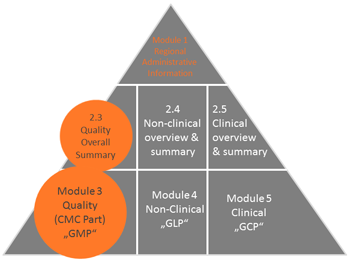
Sciene-based writing of clear technical documentation, e.g., former STED format ("nlVDMAToC" or "IVDMAToC", March 2019) and regulatory documents, i.e., modules 3 and 2.3 of the "Common Technical Documents (CTD)", which are accepted by authorities, e.g., "Drug Master Files, DMFs", "Active Substance Master Files, ASMFs", "Certificates of Suitability, CEP," "Investigational Medicinal Product Dossiers, IMPDs", "Investigational New Drugs, INDs" for national and international registrations (e.g., EU, USA, Canada, Japan), as well as preparation and editing of eCTD according to ICH guidelines M2, M8 with commercially available software.
Under the Mutual Recognition Agreement (MRA), the number of routine FDA inspections in Europe is likely to decrease in the future. However, FDA will continue to perform some "product manufacturing assessment inspections" in recognized countries (currently 27 countries recognized between November 2017 to July 2019), to support marketing approval decisions.
A lot is heared and read about the horrors of FDA inspections and also as many tips about strategies to deal with them are circulating. But, there is no such a thing like the FDA inspection!
FDA inspections depend on the hard facts, as documentary evidence of quality (written evidence), but are influenced as well to a large extent by individual personality characteristics of inspectors and auditees, the so called audit psychology. For this reason it is necessary to develop for each inspected company an individual approach to ensure that the inspection will be a success, i.e., no FDA form 483 observation is filed. In order to apply optimal measures, I have researched in recent years in many actually inspected companies on the complex psychology of the entire inspection phase, from the announcement to the completion of possible reworkings. Typical steps for the preparation and moderration of FDA audits in the field of pharmaceutical and medical technology are:
- Experience-based preparation and optimization of the company and the inspection environment, e.g., accommodation and picking up the inspector(s), catering, room planning, company presentation, front / back office and communication during the inspection
- Training of persons involved, for example, in audit psychology, stress management before / during the audit, goal-oriented presentation of the QM system during the audit
- Review and optimization of the 5 core processes: CAPA, Change Control, Deviations, OOS, Training
 "CAPA --- it's magic" quote of a FDA inspector, May 2015
"CAPA --- it's magic" quote of a FDA inspector, May 2015 - Review and preparation of documents, for example, Quality Manual, Technical / Quality Agreements in accordance with § 9 AMWHV, EU GMP guideline Chapter 7
- Preparation of audits according to FDA's Medical Device Single Audit Program (MDSAP)
Further information:
- Training of internal / external GMP auditors
- Co-Author of the book chapter "Tips for a Successful FDA Inspection" in "The HPLC Expert" [ISBN 978-3-527-33681-4, Wiley-VCH, Weinheim, Ed. Stavros Kromidas, 2016]
- Searchable list of current FDA Warning Letters
Employees or "Human Resources" are the capital of a company. Therefore, employee satisfaction and the environmetal fit of new employees --- regarding internal placement or employment --- are very important for the optimization or development of a company, especially for small and medium-sized enterprises. In this context, I offer the following services:
- Realization of structured workplace and job-requirements analyzes, according to the standards of work and organizational psychology
- Implementation of capability analyzes for internal placement as well as employee search and selection processes with structured interviews (based on the Multimodal Interview MMI, according to Heinz Schuler, 1992), in compliance with the standards DIN 33430 "Requirements for methods and their use in job-related proficiency assessment (own translation)" and ISO 10667 "Assessment service delivery - Procedures and methods to assess people in work and organizational settings"
Training for individuals or groups in the regulated industry. A typical course covers the following topics, which can be customized, e.g., supplemented or shortened:
- Analysis of the working environment, for example, Analysis of the quality management system based on documents, SOPs before the workshop and based on "subjective" information from the personal interview before the training
- The stress components (perceived stressors) and competencies; the 3 stress components and the 3 pillars of stress management, according to Prof. Dr. Gert
Kaluza:
(1) Regenerative Stress Management (recuperate, but correctly)
(2) Mental Stress Management (discover your stress reinforcing paradigms)
(3) Instrumental Stress Management (optimize your work environment ) - Dealing with conflicts in the organization / department, analysis of communication
- Perception of self-image vs. public image ("Johari Window")
- Analysis of the motivations concerning the "work domaine". Strategies and motivation techniques for dealing with job requirements; The experiences in the various "domains" influence each other by "stress spillover", i.e., the transfer of stress between different domains, such as job and private life (intra-individual).
- Learning techniques that enable a better perceived Work - Life - Balance.
The necessary qualifications are available that allow a co-payment / reimbursement by German health insurances according to § 20 SGB V (German Social Security Code).
In order to be able to optimize and to develop systems and processes, you need "evaluable" information from experts of these systems and processes. Laboratory talks, informal networking or individual opinions of individual, particularly communicative employees or customers are not to be regarded as "evaluable" information.
With the help of anonymous employee or customer surveys that are standardized and statistically evaluable (online and / or paper based), questions that are specifically developed for the company about the typical processes, as well as exploratory free-text boxes that can provide representative indications of previously unknown trends in the employee or customer opinion. A survey comprises the following steps:
- Project and target definition
- Development of a experience-based survey design, consultation with the sponsor
- Conduct the survey, statistical analysis of the survey data
- Preparation of a result report
- Feedback of results, presentation of the results on the spot
- Discussion of measures for optimization and development
- Evaluation of the achievement of project and target definition
For further information or to arrange an appointment, please contact me.

Figure: In my projects, I am working according to a comprehensive, "transactional" quality management approach
that comprises the socio-cultural perspective as well as the technical perspective.
©create skills | c/ Alcalá 62 | 29130 Alhaurín de la Torre - Málaga, Spain | info[at]create-skills.de | phone +49 (0)172 62 32 378 |
|
Imprint | Data Privacy Statement |
last update:
2026-05-18
| ![]()